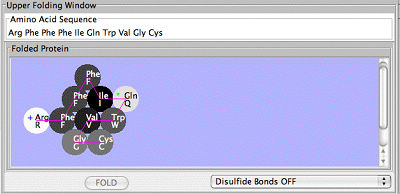
The Protein Investigator gives students an opportunity to interactively explore protein structure and function in a simplified system. This is a highly-simplified model of protein folding. It is not intended to predict the correct structures of any proteins; it is designed to illustrate the major principles involved in that process. The important features of proteins that this software retains are as follows:
Amino acids have side-chains of varying hydrophobicity, charge, and hydrogen bonding capacity.
The amino acids a reconnected in an un-branched chain that can bend.
Hydrophobic amino acids will tend to avoid the water that surrounds the protein; hydrophilic amino acids will bind to the water.
Amino acids that can form hydrogen bonds will tend to form hydrogen bonds if they can.
Positively-charged amino acids will tend to form ionic bonds with negatively-charged amino acids if they can.
Like-charged amino acids will repel each other if they can.
Ionic interactions are stronger than hydrogen bonds, which are stronger than hydrophobic interactions.
Proteins can be folded under oxidizing (Disulfide Bonds ON) or reducing (Disulfide Bonds OFF) conditions.